PMS = Postmarketing Surveillance / Tıbbi Cihazların Piyasa Sonrası Gözetimi
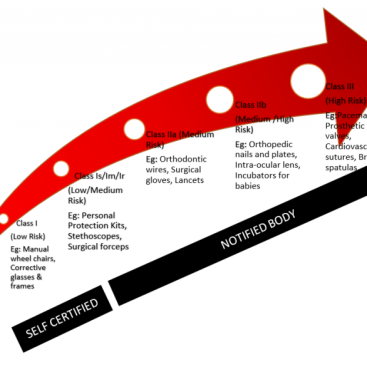
Tıbbi cihazın sınıflandırması ne olursa olsun, Piyasa Sonrası Gözetim gerekli ve üreticilerin bir yükümlülüğüdür. Ek IX ila XI’de tanımlandığı gibi bir CE işareti almak için uygunluk değerlendirme süreçleri, üreticilerin cihazın risk sınıfı ve tipiyle orantılı bir PMS mekanizması oluşturmasını ve sürdürmesini gerektirir, ancak risk sınıfına göre yalnızca gereksinimler değişir.
MDR Madde 2, bölüm 60, PMS’yi üreticilerin ithalatçılar, distribütörler, yetkili temsilciler vb. gibi diğer ekonomik operatörlerle birlikte uyguladığı ve yürüttüğü proaktif ve sistematik bir süreç olarak tanımlar; tıbbi cihaz bilgilerine ve performansına uygun olarak düzeltici ve önleyici faaliyet (DÖF) almak.
Tıbbi cihazları içeren olayların piyasaya arz sonrası gözetimi ve raporlanması, tıbbi cihazların tasarımı, üretimi veya kullanımı ile ilgili sorunların tanımlanmasına olanak tanır ve nihayetinde hasta güvenliğini artırır. Piyasaya arz sonrası gözetim sisteminin amacı, bir cihazın tüm tıbbi cihaz ömrü boyunca kalitesi, performansı ve güvenliği ile ilgili verileri aktif ve sistematik olarak toplamak, kaydetmek ve analiz etmektir.
Bu, üreticilerin risk-fayda değerlendirmesini sürekli olarak güncellemelerine ve gerekli önlemleri gecikmeden başlatmalarına olanak tanır. Üreticiler, tıbbi cihazları ve ilgili cihazları ile ilgili tüm bilgileri rakiplerinden toplamak ve değerlendirmekle yükümlüdür.
Tıbbi cihaz üreticileri, piyasaya arz sonrası gözetim sistemlerini, teknik dokümantasyona dahil edilmesi ve 2017/745 PMS kriterlerine uygunluğunu göstermesi gereken bir PMS Planına (Madde 84) dayandırmalıdır. Ek III, piyasa sonrası verilerin toplanmasını ve kullanılmasını ele alması gereken bir PMS stratejisinin standartlarını ve özünü ortaya koymaktadır.
Tıbbi Cihaz Piyasa Sonrası Gözetim Gereklilikleri
Bir PMS geliştirirken, ürün teknolojisini benzer cihazların üreticisi ve pazarı ile bağlantılı olarak değerlendirmek esastır. Yeni teknoloji üreticileri arasında hasta popülasyonu ve cihaz durumunun karmaşıklığı konusunda farkındalık eksikliği olabilir.
Genel Güvenlik ve Performans Gereksinimleri (GSPR’ler), cihazı piyasaya sunan üretici tarafından PMS toplama ve çıktısına yanıt olarak güncellenmelidir.
Tıbbi Cihaz Piyasa Sonrası Gözetim Raporu
Sınıf I tıbbi cihaz üreticileri (Is, Im ve Ir sınıfları dahil), PMS planında belirtildiği gibi elde edilen verilerin sonuçlarını ve sonuçlarını özetleyen bir piyasaya arz sonrası gözetim raporu sunmakla yükümlüdür. Rapor, alınan önlemlerin gerekçesi ile uygulanan önleyici ve iyileştirici önlemlerin bir özetini içermeli ve gerektiğinde güncellenmelidir.
Piyasa Sonrası Gözetim Raporu veya PMSR, PMS Planına göre yürütülen tüm PMS faaliyetlerinin bir kaydına sahiptir.
Tıbbi Cihaz üreticileri, yöntemleri analiz etmek ve bir Pazar sonrası gözetim planı tasarlamak için diğer departmanlar ve düzenleyici ekiple işbirliği yapacak olan çapraz işlevli bir PMS ekibi oluşturmalıdır. Üreticiler, benim gibi danışmanlar ve düzenleyici uzmanların yardımıyla hedef demografik bilgileri daha kolay belirleyebilir ve tehlikeleri öngörebilir. Aynı şekilde, yeni bir teknoloji piyasaya sürüldüğünde, sorunların erken tespitini sağlamak için etkin bir izleme programı oluşturulmalıdır.
- Üreticileri PMS’yi yürütmede yönlendirir
- Piyasa Sonrası Gözetim planının hazırlanmasında destekler;
- PMS faaliyetlerinin yürütülmesinde kılavuzlar;
- İyi yapılandırılmış PMS planı, prosedürü ve rapor şablonları sağlar;
- PMS verilerinden PMCF gereksinimlerini tanımlar;
- Piyasa Sonrası Gözetim Verilerini analiz eder ve uygun sonuçlar çıkarır.
Piyasa Gözetimi Sonrası Gereksinimleri şu şekilde karşılanabilir:
- Benzer cihazlar için mevcut piyasa verileri , uzun bir kullanım geçmişine sahip düşük riskli cihazlar için klinik değerlendirme gereksinimlerini karşılamak için yeterli olabilir .
- Klinik değerlendirme kriterlerini desteklemek için hakemli literatürden cihazın güvenliği ve performansı hakkında uygun ve ölçülebilir klinik veriler.
- Geliştirme sürecinde beklenmeyen tehlikelerin erken tespitini garanti altına almak için artan izleme.
- Teknolojinin yeterli gerçek dünya klinik kullanımını toplamak için piyasaya arz sonrası klinik takip ( PMCF ) gerekli olabilir.
- Verileri elde etmek için kapsamlı ve proaktif bir strateji (a) noktasında belirtilmiştir. Yöntem, cihazın performansının güvenilir bir şekilde değerlendirilmesine ve piyasadaki benzer ürünlerle karşılaştırmalara izin vermelidir.
- Toplanan verileri değerlendirmek için verimli ve uygun yöntemler ve süreçler;
- Ek I Bölüm 3’te açıklanan fayda-risk analizi ve risk yönetiminin sürekli değerlendirilmesinde kullanım için uygun göstergeler ve eşik değerler;
- Olayların sıklığında veya ciddiyetinde istatistiksel olarak önemli herhangi bir artışın ve ayrıca gözlem süresinin belirlenmesine yönelik teknikler ve süreçler de dahil olmak üzere , 88. Maddenin eğilim raporunda belirtilen olayların yönetimine yönelik teknikler ve protokoller ;
- Yetkili makamlar , onaylanmış kuruluşlar, işletme operatörleri ve kullanıcı ile iletişim için teknikler ve protokoller
- Üreticinin 83, 84 ve 86. maddeler kapsamındaki yükümlülüklerini yerine getirme prosedürlerinin açıklaması.
- Düzeltici faaliyetler gibi uygun önlemleri belirleme ve uygulamaya koyma süreçleri ;
- Tamir edilmesi gerekebilecek cihazları izlemek ve tespit etmek için etkili yöntemler ;
- Ek XIV Bölüm B’de belirtildiği gibi bir PMCF planı veya bir PMCF’nin neden gerekli olmadığına dair bir neden.
MDR’nin gelişiyle karşılaştırıldığında MDD, PMS gerekliliklerinde ISO 14971 madde 9’un gerekliliklerine karşı daha fazla netlik buldu. MDR, 2. Madde (60) uyarınca tüm cihaz üreticilerinin genel görevlerinden biri olarak Piyasa Sonrası Gözetim’i tanımlar. MDR ayrıca, mevzuata uygunluğun izlenmesi ve doğrulanmasından sorumlu çalışanlardan açıkça talepte bulunmuştur. EU MDR ile uyumlu olarak, üreticilerin artık PMS sistemlerini eksiksiz ve metodik bir şekilde proaktif olarak yükseltmeleri gerekiyor.
Bu, üreticilerin, cihaz tipine uygun etkileşimli bir düzeltici eylem veya önleyici eylem yaklaşımı uygulamasına ve ayrıca klinik değerlendirme belgelerini güncellemesine olanak tanır. PMS gereksinimlerinin, kullanım amacına bağlı olarak öğenin oluşturduğu tehlikeyle orantılı olması gerektiğini hatırlamak çok önemlidir.
Tıbbi Cihaz Piyasa Sonrası Gözetimi ve ISO 13485 İlişkisi
Piyasa Sonrası Gözetim, şirketin Kalite Yönetim Sistemine ( ISO 13485 ) entegre edilmeli ve tüm yaşam döngüsü boyunca cihazın kalitesi, performansı ve güvenliği ile ilgili verileri analiz etmek için birlikte oluşturulmalıdır. Ayrıca bu veriler hakkında sonuçlara varılmasına ve şirketin önleyici ve düzeltici faaliyet sistemine bağlanmasına izin verilmelidir.
PMS ‘Reaktif’ olabilir – bir olay meydana geldikten sonra yanıt verir ve büyük ölçüde veri toplama faaliyetleri oldukları için pasif olarak kabul edilir.
PMS ‘Proaktif’ olabilir – olayları meydana gelmeden önce tahmin etmeye ve azaltmaya yönelik çabalar ve cihazın gerçek dünya performansına ilişkin içgörü ve veri sağladığından aktif olarak kabul edilir.
Üretim sonrası izleme için ISO 14971 madde 9 gerekliliği gibi Piyasa Sonrası Gözetim, amaçlanan kullanıma dayalı olarak cihazla ilişkili riskle orantılı olmalıdır. Netlik eksikliği o zamandan beri, Madde 2 (60)’da PMS’yi tüm üreticilerin genel görevlerinden biri olarak tanımlayan MDR ile giderilmiştir.
Ek olarak, MDR, düzenleyici uyumu sağlamaktan (PRRC) sorumlu personel tarafından PMS sistemlerini AB MDR’ye uygun olarak kapsamlı ve sistematik bir şekilde proaktif olarak güncellemeleri gereken üreticilere özel olarak çağrıda bulundu. Bu, üreticilerin, cihaz tipiyle orantılı etkileşimli bir düzeltici eylem veya önleyici eylem süreci oluşturmasına ve klinik değerlendirme belgelerini güncellemesine olanak tanır.
MDR ayrıca, üreticilerin PMS sistemlerini, mevzuat uyumluluğunun (PRRC) izlenmesinden ve sürdürülmesinden sorumlu kişiler tarafından AB MDR’ye uygun olarak kapsamlı ve sistematik bir şekilde proaktif olarak güncellemeleri gerektiğini açıkça belirtmiştir. Bu, üreticilerin, cihaz tipine uygun etkileşimli bir düzeltici eylem veya önleyici eylem yaklaşımı uygulamasına ve ayrıca klinik değerlendirme belgelerini güncellemesine olanak tanır.
PMS gereksinimlerinin, kullanım amacına bağlı olarak öğenin oluşturduğu tehlikeyle orantılı olması gerektiğini hatırlamak çok önemlidir.
ISO 13485, piyasadaki tüm tıbbi cihazlar için geçerli olduğundan, PMS stratejileri proaktif bir Pazar Sonrası Gözetimi ile birlikte geliştirilmelidir. Uyanıklık bir KYS çıktısı olduğundan, bir cihazın teknik dokümantasyonu içinde piyasaya arz sonrası klinik veri toplama çabalarını düzenlemek faydalıdır.
Sonuç olarak, cihazla ilgili önemli olayları yetkililere bildirmek için üretim sonrası aşamada bir tıbbi cihaz uyarı sistemi oluşturmak, daha önce bildirilmemiş olumsuz ürün bilgilerinin keşfedilmesine yardımcı olabilir. Bu değerlendirme, ölümle veya sağlığın önemli ölçüde bozulmasıyla sonuçlanabilecek durumların gelecekte ortaya çıkmasını önlemede faydalı olabilir.