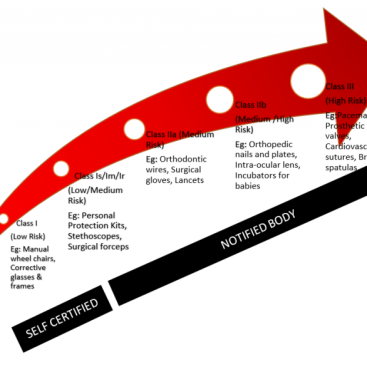
Klinik Değerlendirme Nedir?
- Klinik Değerlendirmede kullanılan doküman ve verilerin kaynakları nedir?
– Literatür taraması ile oluşturulan veriler
– Klinik deneyim ile oluşturulan veriler - Klinik çalışma sonucunda oluşturulan veriler
- Klinik verilerin değerlendirilmesi (Appraisal)Klinik verilerin analiz edilmesi
- Klinik değerlendirme raporunun yazılması
- Risk/Fayda Analizi ve Toplam Kalan Riskin Değerlendirilmesi
- Pazar Sonrası Klinik İzleme ve Pazar Sonrası Takip (Post Market Clinical Follow Up, Post Market Clinical Surveillance)
- Genel hatları ile Medikal Cihazlarda Klinik Çalışma
Temel Tanımlar
MDR Madde 2 (44) klinik değerlendirmeyi, klinik faydalar da dahil olmak üzere, güvenlik ve performansı doğrulamak için bir cihazla ilgili klinik verileri sürekli olarak oluşturmak, toplamak, analiz etmek ve değerlendirmek için sistematik ve planlanmış bir süreç olarak tanımlamaktadır. Tabii ki bu değerlendirme, yasal üretici tarafından beyan edilen kullanım amacı ve klinik bulgular çerçevesinde yapılmalıdır. Buna paralel olarak, Madde 2 (48), klinik veriyi, bir cihazın kullanımıyla oluşturulan ve aşağıdakilerden kaynaklanan güvenlik veya performansla ilgili bilgiler olarak tanımlar. Bu kapsamda klinik veri aşağıda belirtilenlerden elde edilebilir.
- İlgili cihazın klinik araştırması,
- Söz konusu cihazla eşdeğerliği kanıtlanabilen bir cihazın ile yapılan klinik araştırmalar veya bilimsel literatürde bildirilen diğer çalışmalar,
- Söz konusu cihaz veya söz konusu cihazla eşdeğerliğin gösterilebileceği bir cihazla ilgili diğer klinik deneyimler hakkında saygın bilimsel literatürde yayınlanan raporlar,
- Satış sonrası gözetimden ve takipten gelen klinik bulgular.
Bu iki tanım, klinik kanıtları açıklamak ve tıbbi cihaz üreticilerine net bir hedef vermek için Madde 2 (55) ‘de birleştirilmiştir. Dolayısıyla, bir klinik değerlendirme veya klinik araştırma sürecinin amacı, tıbbi cihazın güvenliği hakkında yeterli klinik kanıt oluşturmak olacaktır. MDR’ın temel gereklilikleri ile bazı yöntemsel farklar içermesine rağmen, MDR’dan hemen önce yayınlanan Meddev 2.7.1 Rev 4’ün aynı hedefi vurguladığını belirtmek faydalı olacaktır.
Yukarıda bahsettiğimiz temel tanımlar dışında, MDR Klinik Değerlendirme Gerekliliklerini daha iyi anlamak için Madde 2 (45) ila (57) arasında verilen tanımları incelemenizi tavsiye ederiz.
Madde 2 (55) – Klinik Kanıt: Cihazın güvenli olup olmadığına ve uygun şekilde kullanıldığında amaçlanan klinik yarar(lar)ı elde edip etmediğine ilişkin nitelikli bir değerlendirmeye izin vermek için yeterli miktarda ve kalitede bir cihaza ilişkin klinik veriler ve klinik değerlendirme sonuçları üretici tarafından tasarlanmıştır.MDR Klinik Değerlendirme Gereklilikleri
MDR Clinical Assessment Requirements
Temel terimleri ve tanımları inceledikten sonra, MDR Klinik Değerlendirme Gereksinimlerinin ayrıntılarına bir göz atalım. Yukarıdaki paragrafta belirtildiği gibi, Bölüm VI, klinik değerlendirme ve araştırma için gereksinimleri listelemektedir. Üreticinin CE İşareti için ana görevi, Ek I’de listelenen geçerli genel güvenlik ve performans gerekliliklerine (GSPR) cevap vermektir. 61. Maddeye göre, bu cevaplar, tıbbi cihaz risk sınıfından bağımsız olarak, yeterli klinik kanıt sağlayan klinik verilere dayanmalıdır. Diğer bir deyişle, imalatçı, geçerli GSPR ile uyumluluğu göstermek için yeterli seviyede klinik kanıt sunmalıdır. Süreç, Ek XIV Bölüm A’ya paralel olarak yapılandırılmış bir plan ile yürütülmelidir. Doğal olarak, sürecin çıktısı klinik değerlendirme raporu olacaktır.
Belirli tip Sınıf IIb, implante edilebilir ve Sınıf III cihazlar için ek şartlar, klinik değerlendirme sürecini diğerlerine göre biraz daha karmaşık hale getirmektedir. Asıl sorun, bu cihazlar için aşağıda listelenen belli şartlar yerine getirilemiyor ise, klinik araştırmanın gerekli olmasıdır;
- Cihaz, aynı üretici tarafından halihazırda CE Markalı olarak piyasaya sürülen bir cihazın benzeri ise (değişik bir tipi),
- Değiştirilen cihazın Ek XIV Bölüm 3’e uygun olarak, üretici tarafından pazarlanan cihaza eşdeğer olduğu kanıtlanmış ve bu durum onaylanmış kuruluş tarafından onaylanmış ise,
- Pazarlanan cihazın klinik değerlendirmesi, değiştirilmiş cihazın ilgili güvenlik ve performans gerekliliklerine uygunluğunu kanıtlamaya yeterli ise.
Buna ek olarak, benzer bir cihazın klinik verileri değerlendirme raporunda kullanılabiliyorsa, yasal üretici yukarıda belirtilen tipteki cihazlar için klinik bir araştırma yapmadan bir klinik değerlendirme yapabilir. Her ikisi de kardeş şirket olmadıkça, benzer cihazın üreticisi ile bu tip bir “sözleşme” yapmanın zorluğu, bu opsiyonu çoğunlukla imkansınız kılacaktır. Bu yüzden üreticileri bu tip cihazlar için geçiş döneminde etkin bir satış sonrası klinik takip programı (PMCF) ile kendi verilerini toplamalarını şiddetle tavsiye ediyoruz.
Ayrıca, 93/42 / EEC veya 90/385 / EEC direktifleri kapsamında yeterli bir klinik değerlendirme raporu sunabilen implante edilebilir ve Sınıf III cihaz üreticileri, MDR Madde 61’de listelenen klinik araştırma gerekliliklerinin dışında tutulur. Bu kapsamada kullanılabilecek klinik veriler için MDCG’nin 2020-6 numaralı rehber dokümanını inceleyebilirsiniz.
Öte yandan sutürler, zımbalar, diş dolguları, diş telleri, diş kronları, vidalar, plakalar, teller, pimler, klipsler veya konektörler gibi ilgili maddede listelenen tüm implante edilebilir cihazlar da klinik araştırma gerekliliklerinin dışında tutulmuştur. Lütfen AB komisyonun zaman içerisinde bu listeyi değiştirme hakkını saklı tuttuğunu unutmayın.
61. Maddenin 9. Paragrafı, tıbbi amacı olmayan ancak MDR Ek XVI’ da listelenen cihazlar için gereksinimleri tanımlamaktadır. Bu cihazlar için, klinik yararın gösterilmesi, cihazın performansının beyan edilen kullanım amacına yönelik olarak anlaşılmalıdır. Bu tıbbi cihazlar için klinik değerlendirmeler, piyasaya arz sonrası gözetim, PMCF ve uygun olduğu durumlarda spesifik klinik araştırmalardan elde edilen veriler dahil olmak üzere güvenlikle ilgili verilere dayandırılmalıdır.
Üreticiler, MDR kapsamında klinik değerlendirmenin sürekli güncellenmesi gereken bir süreç olduğunu da unutmamalıdır. Bu süreç, tıbbi cihazın yaşam döngüsü boyunca değişen şartlara göre ele alınmalıdır. Özellikle implante edilebilir ve Sınıf III cihazlar için klinik değerlendirme raporları en az yılda bir kez güncellenmelidir beklemeye alınarak ve ek bilgiler talep edilecektir.
Klinik Değerlendirme Planı
Üreticiler, MDR Klinik Değerlendirme Gerekliliklerine uymak için etkin bir plan hazırlamalıdırlar. Bu plan, Ek XIV Bölüm A’da listelenen gereksinimlere uygun olmalıdır.
- İlgili klinik veriler ile desteklenmesi gereken genel güvenlik ve performans gereksinimlerinin tanımı;
- Cihazın beyan edilen kullanım amacı;
- Açık endikasyonlar ve kontrendikasyonlar ile amaçlanan hedef grupların net bir tanımı;
- İlgili ve belirlenmiş klinik sonuç parametreleri dahil hastalara yönelik amaçlanan klinik faydaların ayrıntılı bir açıklaması;
- Kalan risklerin ve yan etkilerin belirlenmesine yönelik klinik güvenliğin kalitatif ve kantitatif yönlerinin incelenmesi için kullanılacak yöntemler;
- Tıptaki en son teknolojiye dayalı olarak, çeşitli endikasyonlar için ve cihazın beyan edilen kullanım amacı için fayda-risk oranının kabul edilebilirliğini belirlemek için kullanılacak parametrelerin bir listesi;
- Farmakolojik, cansız hayvan veya insan dokularının kullanımı gibi belirli bileşenlerle ilgili fayda-risk konularının nasıl ele alınacağına dair bir gösterge; ve
- İlk hasta çalışmaları, fizibilite ve pilot çalışmalar gibi temel klinik araştırmalardan doğrulayıcı araştırmalara ilerlemeyi gösteren bir klinik geliştirme planı ve Ek XIV Bölüm B’de belirtildiği gibi bir endikasyonla birlikte bir PMCF kilometre taşları ve potansiyel kabul kriterlerinin açıklaması.
Klinik değerlendirmenizin diğer herhangi bir aşamasında olduğu gibi, planlama aşamasında MDCG rehber dokümanı MDCG 2020 -13’ten yararlanabilirsiniz. Bu kılavuz, onaylanmış kuruluşlara (NB) yönelik olsa da üretici olarak sürecinizi bu belgede verilen örneklere paralel olarak yapılandırmak denetim sırasında büyük faydalar sağlayacaktır.
Literatür Tarama Protokolü
Klinik değerlendirme süreci sırasında, üreticiler tıbbi cihazlarıyla ilgili mevcut klinik verileri tanımlamalıdırlar. Etkili bir literatür araştırması için, Meddev 2.7.1 Rev 4 Ek A.5’e başvurabilirsiniz. MEDLINE veya PubMed gibi bilimsel literatür veri tabanlarının araştırılmasına dayalı protokolü iyi bir başlangıç noktası olacaktır. Veri tabanları, doğru anahtar kelimeler kullanılarak benzer amaçlara sahip eşdeğer cihazlar (Teknik, Biyolojik, Klinik) için taranmalıdır. Klinik kanıtlardaki boşluklar, sistematik bir literatür taraması yoluyla tespit edilerek, klinik değerlendirme raporu için bir girdi olarak rapor edilecektir.
Klinik Değerlendirme Raporu
Yukarıda belirtildiği gibi, MDCG 2020-13’ün şablon olarak kullanılması, sorunsuz bir belgelendirme süreci sağlamak için çok iyi bir çözüm olabilir. Üreticiler ayrıca Meddev 2.7.1 Rev 4’ün MDR Klinik değerlendirme gereksinimleriyle çelişmeyen bölümlerinden de rapor formatını belirlemek için yararlanabilir.
Başarılı bir klinik değerlendirme raporu için kilit nokta, değerlendirme planına göre literatür inceleme protokolünün uygulanmasıyla üretilen klinik verilerin değerlendirilmesidir. IMDRF rehber dokümanı WG / N56FINAL: 2019, Ek F’de örnek değerlendirme yöntemleri bulunmaktadır.
Klinik değerlendirmeyle ilgili belirli konularda daha fazla açıklamaya veya yüksek riskli tıbbi cihazlarınızın klinik araştırma gereksinimleri konusunda yardıma ihtiyacınız varsa, bizimle iletişime geçmekten çekinmeyin.