PMS = Postmarketing Surveillance
Regardless of the classification of the medical device, Post-Market Inspection is necessary and a manufacturer’s obligation. Conformity assessment processes for obtaining a CE marking as defined in Annexes IX to XI require manufacturers to establish and maintain a PMS mechanism commensurate with the risk class and type of device, but only the requirements vary by risk class.
MDR Clause 2, part 60, PMS is not permitted by manufacturers to importers, distributors, authorized representatives, etc. It defines it as a proactive and systematic process that it implements and executes together with other economic operators such as; To take corrective and preventive action (CAPA) in accordance with medical device information and performance.
Post-market surveillance and reporting of incidents involving medical devices allows for the identification of issues related to the design, manufacture or use of medical devices and ultimately improves patient safety. The purpose of the post-market surveillance system is to actively and systematically collect, record and analyze data on the quality, performance and safety of a device over the entire medical device lifecycle.
This allows manufacturers to continuously update the risk-benefit assessment and initiate the necessary measures without delay. Manufacturers are obliged to collect and evaluate all information about their medical devices and related devices from their competitors.
Medical device manufacturers should base their post-market surveillance system on a PMS Plan (Article 84), which must be included in the technical documentation and demonstrate compliance with the 2017/745 PMS criteria. Annex III sets out the standards and substance of a PMS strategy that should address the collection and use of post-market data.
Medical Device Post-Market Surveillance Requirements
When developing a PMS, it is essential to evaluate the product technology in relation to the manufacturer and market of similar devices. There may be a lack of awareness of the patient population and complexity of device status among new technology manufacturers.
General Safety and Performance Requirements (GSPRs) must be updated in response to PMS collection and output by the device manufacturer.
Medical Device Post-Market Inspection Report
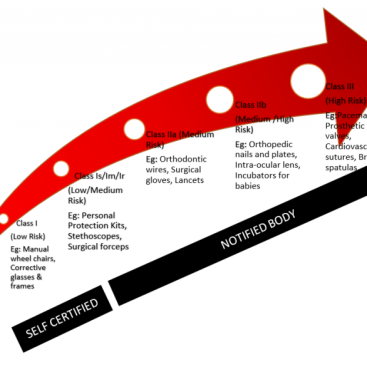
Manufacturers of class I medical devices (including classes Is, Im and Ir) are required to submit a post-market surveillance report summarizing the results and conclusions of the data obtained as specified in the PMS plan. The report should contain the rationale for the measures taken and a summary of the preventive and remedial measures implemented and should be updated as necessary.
The Post-Market Surveillance Report or PMSR has a record of all PMS activities carried out according to the PMS Plan.
Medical Device manufacturers should create a cross-functional PMS team that will collaborate with other departments and the regulatory team to analyze methods and design a post-market surveillance plan. Manufacturers can more easily identify target demographics and anticipate hazards with the help of consultants and regulatory experts like myself. Likewise, whenever a new technology is released, an effective monitoring program should be established to ensure early detection of problems.
- Guides manufacturers in executing PMS
- Supports in the preparation of the Post-Market Surveillance plan;
- Guidelines in the conduct of PMS activities;
- Provides well-structured PMS plan, procedure and report templates;
- Defines PMCF requirements from PMS data;
- Analyzes Post Market Surveillance Data and draws appropriate conclusions.
Post Market Surveillance Requirements can be met by:
- Existing market data for similar devices may be sufficient to meet clinical evaluation requirements for low-risk devices with a long history of use.
- Appropriate and measurable clinical data on device safety and performance from peer-reviewed literature to support clinical evaluation criteria.
- Increased monitoring to ensure early detection of unexpected hazards during development.
- Post-market clinical follow-up (PMCF) may be necessary to gather sufficient real-world clinical use of the technology.
- A comprehensive and proactive strategy for obtaining the data is outlined in point (a). The method should allow reliable evaluation of the device’s performance and comparisons with similar products on the market.
- Efficient and appropriate methods and processes for evaluating the data collected;
- Appropriate indicators and thresholds for use in the ongoing assessment of benefit-risk analysis and risk management as described in Annex I Section 3;
- Techniques and protocols for the management of events as set forth in the trend report of Article 88, including techniques and processes for determining any statistically significant increase in the frequency or severity of events, as well as the duration of observation;
- Techniques and protocols for communication with competent authorities, notified bodies, business operators and the user
- Description of the manufacturer’s procedures for fulfilling its obligations under Articles 83, 84 and 86.
- Processes to identify and implement appropriate measures, such as corrective actions;
- Effective methods for monitoring and detecting devices that may need repair;
- A PMCF plan as set out in Annex XIV Part B or a reason why a PMCF is not required.
Compared to the advent of MDR, MDD found greater clarity in its PMS requirements versus the requirements of ISO 14971 clause 9. The MDR defines Post-Market Inspection as one of the general duties of all device manufacturers pursuant to Article 2 (60). MDR has also made explicit requests to employees responsible for monitoring and verifying regulatory compliance. In line with EU MDR, manufacturers now need to proactively upgrade their PMS systems in a thorough and methodical way.
This allows manufacturers to implement an interactive corrective or preventive action approach appropriate to the device type, as well as to update clinical evaluation documentation. It is very important to remember that PMS requirements must be proportional to the hazard posed by the item based on its intended use.
Medical Device Post-Market Inspection and ISO 13485 Relationship
Post-Market Inspection should be integrated into the company’s Quality Management System (ISO 13485) and co-created to analyze data on device quality, performance and safety throughout its entire lifecycle. It should also be allowed to draw conclusions about this data and link it to the company’s system of preventive and corrective action.
PMS can be ‘Reactive’ – responds after an event has occurred and are largely considered passive as they are data collection activities.
PMS can be ‘Proactive’ – Efforts to predict and mitigate events before they occur are considered active as they provide insight and data on the device’s real-world performance.
Like the ISO 14971 clause 9 requirement for post-production monitoring, Post-Market Surveillance should be commensurate with the risk associated with the device based on the intended use. The lack of clarity has since been rectified by the MDR, which in Article 2 (60) defines PMS as one of the general duties of all manufacturers.
In addition, the MDR specifically called on manufacturers to be required to proactively update their PMS systems comprehensively and systematically in accordance with the EU MDR by staff responsible for ensuring regulatory compliance (PRRC). This allows manufacturers to create an interactive corrective or preventive action process commensurate with device type and update clinical evaluation documentation.
The MDR has also made it clear that manufacturers must proactively update their PMS systems in a comprehensive and systematic manner in line with the EU MDR by those responsible for monitoring and maintaining regulatory compliance (PRRC). This allows manufacturers to implement an interactive corrective or preventive action approach appropriate to the device type, as well as to update clinical evaluation documentation.
It is very important to remember that PMS requirements must be proportional to the hazard posed by the item based on its intended use.
As ISO 13485 applies to all medical devices on the market, PMS strategies should be developed in conjunction with a proactive Post-Market Inspection. Because alertness is a QMS output, it is useful to organize post-market clinical data collection efforts within a device’s technical documentation.
As a result, establishing a post-production medical device alert system to notify authorities of significant device-related events can help discover previously unreported adverse product information. This assessment can be useful in preventing future conditions that could result in death or significant impairment of health.