What is Clinical Evaluation?
- What are the sources of documents and data used in Clinical Evaluation?
– Data generated by literature review
– Data generated by clinical experience - Data generated as a result of clinical study
- Evaluation of clinical data (Appraisal)Analysis of clinical data
- Writing the clinical evaluation report
- Risk/Benefit Analysis and Evaluation of Total Residual Risk
- Post Market Clinical Follow Up, Post Market Clinical Surveillance
- Clinical Study in Medical Devices with General Lines
Basic Definitions
MDR Article 2 (44) defines clinical evaluation as a systematic and planned process to continually generate, collect, analyze and evaluate clinical data on a device to confirm safety and performance, including clinical benefits. Of course, this evaluation should be made within the framework of the intended use and clinical findings declared by the legal manufacturer. Accordingly, Article 2 (48) defines clinical data as safety or performance-related information generated by the use of a device resulting from: In this context, clinical data can be obtained from the following.
- Clinical research of the relevant device,
- Clinical studies or other studies reported in the scientific literature with a device that can be proven to be equivalent to the device in question,
- Reports published in the reputable scientific literature on the device in question or other clinical experience with a device for which equivalence can be demonstrated;
- Clinical findings from postmarketing surveillance and follow-up.
These two definitions are combined in Article 2 (55) to describe the clinical evidence and give medical device manufacturers a clear target. Thus, the purpose of a clinical evaluation or clinical trial process will be to establish sufficient clinical evidence about the safety of the medical device. It is worth noting that Meddev 2.7.1 Rev 4, published just before the MDR, emphasizes the same goal, although it contains some methodological differences with the core requirements of the MDR.
Apart from the basic definitions we mentioned above, we recommend that you review the definitions given in Items 2 (45) to (57) to better understand the MDR Clinical Assessment Requirements.
Clause 2 (55) – Clinical Evidence: Clinical data and clinical evaluation results for a device of sufficient quantity and quality have been designed by the manufacturer to permit a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s) when used appropriately. .
MDR Clinical Assessment Requirements
After reviewing the key terms and definitions, let’s take a look at the details of the MDR Clinical Assessment Requirements. As noted in the paragraph above, Chapter VI lists the requirements for clinical evaluation and research. The main task of the manufacturer for CE Marking is to respond to the applicable general safety and performance requirements (GSPR) listed in Annex I. According to Article 61, these answers should be based on clinical data providing sufficient clinical evidence, regardless of the medical device risk class. In other words, the manufacturer must provide sufficient clinical evidence to demonstrate compliance with the applicable GSPR. The process should be carried out with a structured plan in line with Annex XIV Part A. Naturally, the output of the process will be the clinical evaluation report.
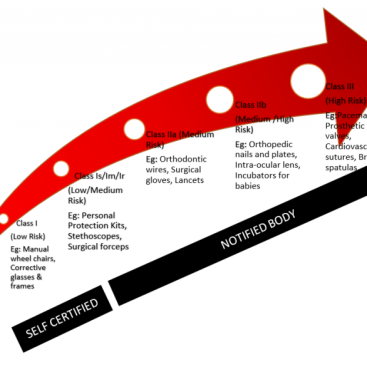
Additional requirements for certain types of Class IIb, implantable, and Class III devices complicate the clinical evaluation process over others. The main problem is that clinical investigation is required for these devices if certain requirements listed below cannot be met;
- If the device is similar to a device already released as CE Marked by the same manufacturer (a different type),
- If the replacement device has been proven to be equivalent to the device marketed by the manufacturer in accordance with Annex XIV Part 3 and this has been approved by the notified body,
- The clinical evaluation of the marketed device is sufficient to demonstrate compliance of the modified device with the relevant safety and performance requirements.
In addition, if the clinical data of a similar device can be used in the evaluation report, the legal manufacturer may conduct a clinical evaluation for the above-mentioned types of devices without a clinical investigation. Unless both are sister companies, the difficulty of entering into this type of “contract” with the manufacturer of a similar device will often make this option unfeasible. Therefore, we strongly recommend that manufacturers collect their own data for this type of device with an effective post-market clinical follow-up program (PMCF) during the transition period.
In addition, manufacturers of implantable and Class III devices capable of providing an adequate clinical evaluation report under the 93/42/EEC or 90/385/EEC directives are excluded from the clinical investigation requirements listed in MDR Article 61. For clinical data that can be used in this context, you can review MDCG’s guide document numbered 2020-6.
On the other hand, all implantable devices listed in the relevant article, such as sutures, staples, dental fillings, braces, dental crowns, screws, plates, wires, pins, clips or connectors, are also excluded from clinical research requirements. Please note that the EU commission reserves the right to change this list over time.
Paragraph 9 of Article 61 defines requirements for devices that do not have a medical purpose but are listed in Annex XVI to MDR. For these devices, demonstration of clinical benefit should be understood as the performance of the device for its declared intended use. Clinical evaluations for these medical devices should be based on safety-related data, including post-market surveillance, PMCF and, where appropriate, data from specific clinical trials.
Manufacturers should also keep in mind that clinical evaluation under MDR is a process that needs to be constantly updated. This process should be addressed according to changing conditions throughout the life cycle of the medical device. In particular, for implantable and Class III devices, clinical evaluation reports should be updated at least annually, on hold, and additional information will be requested.
Clinical Evaluation Plan
Manufacturers should establish an effective plan to comply with MDR Clinical Evaluation Requirements. This plan must comply with the requirements listed in Annex XIV Part A.
- Description of general safety and performance requirements that should be supported by relevant clinical data;
- The declared purpose of use of the device;
- A clear definition of intended target groups with clear indications and contraindications;
- A detailed description of the intended clinical benefits for patients, including relevant and established clinical outcome parameters;
- Methods for examining the qualitative and quantitative aspects of clinical safety to identify residual risks and adverse effects;
- A list of parameters to be used to determine the acceptability of the benefit-risk ratio for the various indications and for the declared intended use of the device, based on the state of the art in medicine;
- An indication of how to address the benefit-risk issues associated with certain ingredients, such as the use of pharmacological, inanimate or human tissues; and a clinical development plan showing progress from basic clinical trials to confirmatory trials, such as initial patient studies, feasibility, and pilot studies, and a description of PMCF milestones and potential acceptance criteria with an indication as set out in Annex XIV Part B.
As with any other phase of your clinical evaluation, you can benefit from the MDCG guidance document MDCG 2020-13 during the planning phase. Although this guide is aimed at notified bodies (NB), structuring your process as a manufacturer in line with the examples given in this document will provide great benefits during the audit.
Literatür Tarama Protokolü
During the clinical evaluation process, urLiterature Search Protocol users should identify available clinical data on their medical device. For an effective literature search, you can refer to Meddev 2.7.1 Rev 4 Appendix A.5. Its protocol based on searching scientific literature databases such as MEDLINE or PubMed would be a good starting point. Databases should be searched for equivalent devices (Technical, Biological, Clinical) with similar purposes using the correct keywords. Gaps in clinical evidence will be identified through a systematic literature review and reported as an input for the clinical evaluation report.