Our Scope of Consultancy
General
- Consults with customers and determines Device Scope for CE Marking along with Models, Variants and Brands.
- Discusses and informs the customers’ team members on the EN ISO 13485:2016 Application.
- It regulates the Organizational Structure with its Duties and Responsibilities.
- Assists in identifying the Notified Body and submitting the MDR/IVDR Application.
Technical
- With the support of the manufacturer, it determines the product-specific standards, harmonized and general standards applicable to the device.
- It collects information about materials, components, imported items, outsourced items, packaging requirement specifications, along with Bill of Materials from Design and Development.
- The production process flow collects details about environmental conditions.
- With the support of the manufacturer, the device collects details about packaging, labeling and shipping.
- It defines the device definition, purpose of use, mechanism of action with the support of the manufacturer.
- Supports security and performance requirements analysis and testing with support from the manufacturer.
- Performs EN ISO 13485:2016 Internal GAP Assessment/IQA.
- Performs technical documentation consolidation and internal review and correction.
- Performs on-site QMS & Product Audit, then closing all findings with manufacturer’s support.
Biological Evaluation
- Identify sources (Internal/External) for Safety Assessment, Biocompatibility Testing, Physical characterization tests, Chemical characterization tests, Stability studies, Validations, Verifications etc. with the support of the manufacturer.
- Supports in biological assessment documentation and testing
Clinical evaluation
- Identifies equivalent/similar device with same risk biological clinical and technical equivalence with manufacturer’s support
- It carries out a detailed assessment of a similar device generation/generation available in the European Union or international markets, given by the manufacturer.
- Identifies patient population, clinical conditions, contraindications, warnings with manufacturer’s support
- Conducts Clinical Evaluation with Post-Market Surveillance and Periodic Safety Update Report
Benefit-Risk Analysis
- Performs Risk Identification and Benefit-Risk Analysis with the support of the manufacturer
- With the support of the manufacturer, Risk Management Plan (RMP), Hazard Traceability Matrix (HTM), Risk Management File (RMF) etc. Creates Risk Management documents.
- Identify and resolve risk analysis gaps
Post-Market Clinical Follow-up
- Supports manufacturer in setting PMS plan and PMS report/PSUR
- Supports the producer in carrying out the Post-Market Clinical Follow-up (PMCF), sets the PMCF plan and related reports
Declaration of Conformity
- It determines the conformity assessment route and draws a Declaration of Conformity.
- Confirms with the manufacturer regarding the details to be added to the document, such as the UDI-DI number, harmonized and harmonized standards, common features, etc.
Notified Body
- Works as an intermediary between the Notified Body and the manufacturer – technical documentation presentation and audit support
- Notified Body corrects review comments, changes/responds to Notified Body with correction and supporting evidence
EU 2017/745 – MDR Consulting
With our CE certification Consultancy service, we help medical device manufacturers commercialize their products in the EU market. As it’s time to switch from MDD to MDR, we can help you identify what’s missing from your technical file and what needs to be added.
By now you might have learned what changes are applied in MDR compared to MDD but if you don’t know anything, don’t worry! As your advisor, we will guide you in the right direction to place your medical device on the EU market.
Here are a few things to consider when migrating to MDR and where we can support:
- Compliance with General Safety and Performance Requirements
- Implementation of Unique Device Identifier (UDI) to track your devices
- Powerful and in-depth clinical data to substantiate safety and performance claims
- Stricter equivalence measures
- Addition of reusable surgical devices requiring new classification rules and NB surveillance
- Reporting of incidents on the EU portal – EUDAMED
EU 2017/746 – IVDR CE Certification Services
With the introduction of IVDR, it turned out that bringing IVD devices to the EU market has turned into a nightmare. As it is a transition period from IVDD to IVDR, we can help you identify what is missing from your technical file and what needs updating.
The main changes you need to consider when sending your IVD device to the EU market and the areas where we can help you with our IVDR CE certification consultancy service are:
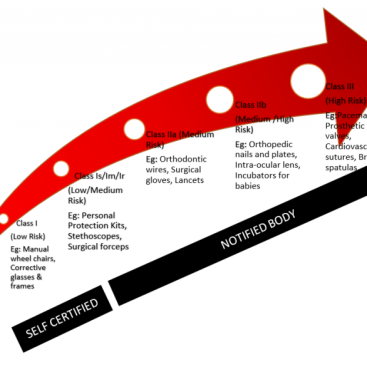
- Risk classes – moved from list-based approach to risk-based approach. The four risk categories are A, B, C and D.
- Conformity assessment routes have been changed – to reflect the new classification rules.
- The Performance Evaluation required during the lifetime of the device should be done according to the Performance Evaluation Plan.
- Scientific validity requires providing clinical evidence reports on analytical performance and clinical performance.
- Post Market Activities – Post Market Performance Report (PMPF) is a new requirement, PMS plan, PMS report and Event report and trend.
- Security and Performance Summary and Unique Device Identifier (UDI) need for C&D classes for inspection and traceability requests.