CE belgesi, Avrupa Birliği (AB) ülkelerinde satılan birçok ürün için zorunlu bir uygunluk işaretidir. Özellikle tıbbi cihazlar ve in vitro tanı cihazları gibi hassas ürünler için CE belgesi alım süreci daha da karmaşık hale gelebilir. Bu noktada devreye CE Belgelendirme, Tıbbi Cihaz Danışmanlığı, MDR (Tıbbi Cihazlar Yönetmeliği) ve IVDR süreci uzmanları girer.
CE Belgelendirme Danışmanlığı Nedir?
CE Belgelendirme Danışmanları, üreticilere ürünlerini AB standartlarına uygun hale getirme konusunda rehberlik eden uzmanlardır. Bu danışmanlar, ürün değerlendirmesinden belge hazırlığına, test ve denetim koordinasyonundan süreç yönetimine kadar bir dizi hizmet sunarak firmalara CE belgesi alım sürecinde yardımcı olurlar.
Tıbbi Cihaz Danışmanlığı Nasıl Yardımcı Olur?
Tıbbi Cihaz Danışmanları, özellikle sağlık sektöründe kullanılan ürünler için CE belgesi alım sürecinde uzmanlaşmıştır. Bu danışmanlar, tıbbi cihazların AB direktifleri ve standartlarına uygunluğunu sağlamak için gerekli adımları belirler ve firmalara uygun belgeleri hazırlamak konusunda rehberlik eder. Aynı zamanda ürünlerin test edilmesi ve denetlenmesi süreçlerini de koordine eder.
MDR ve IVDR Güncellemeleri
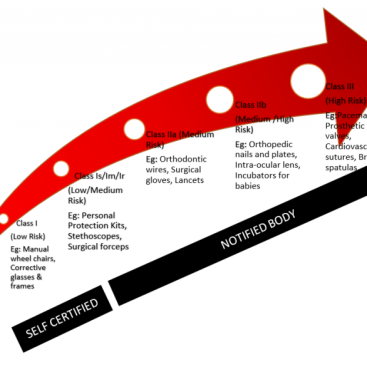
EU 2017/745 MDR, tıbbi cihazlar için, EU 2017/746 IVDR ise in vitro tanı cihazları için AB düzenlemelerini kapsar. Bu yönetmelikler, üreticilere daha fazla sorumluluk getirir ve ürünlerin güvenliği ve etkililiğini sağlamak amacıyla tasarlanmıştır. CE belgesi almak isteyen firmalar için MDR ve IVDR süreçleri önemlidir ve uzman rehberlik gerektirebilir.
Avantajları Nelerdir?
- Uzman Rehberlik: Her iki danışmanlık türü de uzmanlık alanlarında deneyime sahip profesyoneller tarafından sunulur.
- Süreç Yönetimi: CE belgesi alım sürecinin yönetimi, firmaların zaman ve kaynak tasarrufu yapmalarına yardımcı olur.
- Güncel Bilgiler: Danışmanlar, AB yönetmeliklerindeki değişiklikleri sürekli takip eder ve firmaları bu değişiklikler konusunda bilgilendirir.
CE Belgelendirme, Tıbbi Cihaz Danışmanlığı, MDR ve IVDR süreçlerinin birleşimi, üreticilere ürünlerini başarıyla piyasaya sürmeleri konusunda kapsamlı bir destek sunar. Bu hizmetler, hem uygunluk sağlama sürecini kolaylaştırır hem de pazarlama avantajları elde etmelerine yardımcı olur.
Danışmanlık Hizmetlerimizi birkaç başlık altında incelememiz gerekirse;
Genel
- Müşterilerle görüşerek CE İşaretlemesi için Cihaz Kapsamını Modeller, Varyantlar ve Markalar ile birlikte belirler.
- Müşterilerin ekip üyeleri ile EN ISO 13485:2016 Uygulaması hakkında görüşür ve bilgilendirir.
- Organizasyon Yapısını Görev ve Sorumlulukları ile birlikte düzenler.
- Onaylanmış Kuruluşun belirlenmesine ve MDR/IVDR Başvurusunun yapılmasına yardımcı olur.
Teknik
- Üreticinin desteği ile ürüne özel standartları, cihaza uygulanabilir harmonize ve genel standartları belirler.
- Tasarım ve Geliştirmeden Malzeme Listesi ile birlikte malzemeler, bileşenler, ithal kalemler, dış kaynaklı kalemler, paketleme gereksinimi özellikleri hakkında bilgi toplar.
- Üretim süreci akışı, çevresel koşullarla ilgili ayrıntıları toplar.
- Üreticinin desteği ile cihazın ambalajlanması, etiketlenmesi ve sevkiyatı ile ilgili detayları toplar.
- Üreticinin desteği ile cihaz tanımı, kullanım amacı, etki mekanizmasını tanımlar.
- Üreticiden aldığı destek ile güvenlik ve performans gereksinimleri analiz ve testlerini destekler.
- EN ISO 13485:2016 Dahili GAP Değerlendirmesi/IQA gerçekleştirir.
- Teknik dokümantasyon konsolidasyonu ve dahili inceleme ve düzeltme gerçekleştirir.
- Yerinde KYS ve Ürün Denetimi gerçekleştirir, ardından üreticinin desteğiyle tüm bulguları kapatır.
Biyolojik Değerlendirme
- Üreticinin desteği ile Güvenlik Değerlendirmesi, Biyouyumluluk Testi, Fiziksel karakterizasyon testleri, Kimyasal karakterizasyon testleri, Stabilite çalışmaları, Doğrulamalar, Doğrulamalar vb. için kaynakları (Dahili / Harici) belirler.
- Biyolojik değerlendirme dokümantasyonunu ve testlerini destekler
Klinik Değerlendirme
- Üreticinin desteği ile aynı risk biyolojik klinik ve teknik eşdeğerliğe sahip eşdeğer/benzer cihazı belirler.
- Üretici tarafından verilen Avrupa Birliği veya uluslararası pazarlarda mevcut benzer bir cihaz neslinin/neslinin ayrıntılı bir değerlendirmesini yapar.
- Üretici desteği ile hasta popülasyonunu, klinik koşulları, kontrendikasyonları, uyarıları tanımlar.
- Pazarlama Sonrası Gözetim ve Periyodik Güvenlik Güncelleme Raporu ile Klinik Değerlendirme Yürütür.
Fayda-Risk Analizi
- Üreticinin desteği ile Risk Belirleme ve Fayda-Risk Analizi gerçekleştirir.
- Üreticinin desteği ile Risk Yönetim Planı (RYP), Tehlike İzlenebilirlik Matrisi (HTM), Risk Yönetim Dosyası (RMF) vb. Risk Yönetimi dokümanlarını oluşturur.
- Risk analizi boşluklarını belirler ve çözer.
Pazarlama Sonrası Klinik Takip
- PMS planının ve PMS raporunun/PSUR’un oluşturulmasında üreticiyi destekler.
- Pazar Sonrası Klinik Takibin (PMCF) yürütülmesinde üreticiyi destekler, PMCF planını ve ilgili raporları belirler.
Uygunluk Beyanı
- Uygunluk değerlendirme rotasını belirler ve Uygunluk Beyanı çizer.
- UDI-DI numarası, uyumlaştırılmış ve uyumlaştırılacak standartlar, ortak özellikler vb. gibi belgeye eklenecek detaylar konusunda üretici ile teyitleşir.
Onaylanmış Kuruluş
- Onaylanmış Kuruluş ile üretici arasında aracı olarak çalışır – teknik dokümantasyon sunumu ve denetim desteği.
- Onaylanmış Kuruluş denetim yorumlarını düzeltir, düzeltme ve destekleyici kanıtlarla birlikte Onaylanmış Kuruluşa değişiklik yapar/yanıt verir.
AB 2017/745 – MDR Danışmanlık
CE belgelendirme Danışmanlık hizmetimiz ile tıbbi cihaz üreticilerinin ürünlerini AB pazarında ticarileştirmelerine yardımcı oluyoruz. MDD’den MDR’ye geçme zamanı geldiğinde, teknik dosyanızda nelerin eksik olduğunu ve nelerin eklenmesi gerektiğini belirlemenize yardımcı olabiliriz.
Şimdiye kadar MDD’ye kıyasla MDR’de hangi değişikliklerin uygulandığını öğrenmiş olabilirsiniz, ancak hiçbir şey bilmiyorsanız endişelenmeyin! Danışmanınız olarak, tıbbi cihazınızı AB pazarına sunmanız için size doğru yönde rehberlik edeceğiz.
İşte MDR’ye geçerken göz önünde bulundurmanız gereken ve size destek olabileceğimiz birkaç nokta:
- Genel Güvenlik ve Performans Gerekliliklerine Uygunluk
- Cihazlarınızı takip etmek için Benzersiz Cihaz Tanımlayıcısının (UDI) uygulanması
- Güvenlik ve performans iddialarını kanıtlamak için güçlü ve derinlemesine klinik veriler
- Daha katı eşdeğerlik önlemleri
- Yeni sınıflandırma kuralları ve Onaylanmış Kuruluş gözetimi gerektiren yeniden kullanılabilir cerrahi cihazların eklenmesi
- Olayların AB portalında raporlanması – EUDAMED
AB 2017/746 – IVDR CE Belgelendirme Hizmetleri
IVDR’nin yürürlüğe girmesiyle birlikte IVD cihazlarını AB pazarına getirmenin bir kabusa dönüştüğü ortaya çıktı. IVDD’den IVDR’ye geçiş dönemi olduğu için, teknik dosyanızda nelerin eksik olduğunu ve nelerin güncellenmesi gerektiğini belirlemenize yardımcı olabiliriz.
IVD cihazınızı AB pazarına gönderirken göz önünde bulundurmanız gereken başlıca değişiklikler ve IVDR CE belgelendirme danışmanlık hizmetimizle size yardımcı olabileceğimiz alanlar şunlardır:
- Risk sınıfları – liste bazlı yaklaşımdan risk bazlı yaklaşıma geçildi. Dört risk kategorisi A, B, C ve D’dir.
- Uygunluk değerlendirme rotaları – yeni sınıflandırma kurallarını yansıtacak şekilde değiştirilmiştir.
- Cihazın kullanım ömrü boyunca gerekli olan Performans Değerlendirmesi, Performans Değerlendirme Planına göre yapılmalıdır.
- Bilimsel geçerlilik, analitik performans ve klinik performans hakkında klinik kanıt raporlarının sağlanmasını gerektirir.
- Pazar Sonrası Faaliyetler – Pazar Sonrası Performans Raporu (PMPF) yeni bir gerekliliktir, PMS planı, PMS raporu ve Olay raporu ve trendi.
- Denetim ve izlenebilirlik talepleri için C&D sınıfları için Güvenlik ve Performans Özeti ve Benzersiz Cihaz Tanımlayıcı (UDI) ihtiyacı.